
java -jar mib.jar input_option [processing_options]
-f file_name
where the file_name is a file containing set of SMILESes, or SDfile. SDfile will be automatically recognized by an extension ".sdf", ".sd" or ".mol".
All other files are assumed to contain SMILES. In this case SMILES must be a first item in a line.
The line may contain also other items (molecule name, other data), tab separated.
Compressed files with extension .gz, gzip, or zip may be processed without necessity to unpack them.
Data from the SDfile may be retrieved by using the -keep parameter. For example:
java -jar mib.jar -f mdpi.smi -keep "MolName,Amount"
retrieves also parameters named MolName and Amount from the SDfile.
If you want to retrieve all parameters use the parameter -keepall
Processing of a single SMILES is possible by using a -smi input parameter (in this case SMILES string must be in quotes).
java -jar mib.jar -smi 'molecule_smiles'
When no output options are provided, a file with canonised SMILES is generated and sent to the standard output.
When a SDfile should be generated, the parameters -out sdf is required.
-nostereo stereo information in molecules will not be considerer
-normalizeCharges atomic charges will be normalized when possible
-singlePart only the main (largest) part of a multipart molecule will be processed
-standardize is a shortcut for all three previous options together
-normalizeIsotopes removes all isotope labels from atoms
-isostandardize is a shortcut for -standardize plus -normaliseIsotopes
The mib package performs strict valence checking and discards molecules violating organic valence rules (when skipping molecules with errors, respective error messages are issued). They keyword:
-kmwve (mnemonic for "keep molecules with valence errors") allows processing also of molecules with non-standard valencies
-properties
The following properties are available on the output (in this order); items are tab separated:
logP octanol-water partition coefficient
PSA polar surface area
number of nonhydrogen atoms
molecular weight
number of hydrogen-bond acceptors (O and N atoms)
number of hydrogen-bond donors (OH and NH groups)
number of Rule of 5 violations
number of rotatable bonds
molecular volume
When using the keyword -header in the SMILES output mode, the first line of output is a header with property names.
The mib property calculation engine is used in numerous instances by our industry customers and powers also our free online property calculation tool.
-formula the molecule formula is part of the output. This keyword may be used together with the -properties keyword.
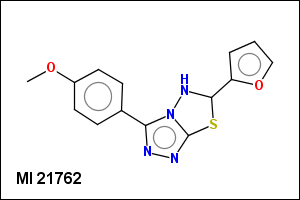
mib allows fragmentation of molecules into various types of fragments. Below examples of various fragmentation options are given. The parent molecule used for the fragmentation is the structure shown above.
-r1 - substituents (Rgroups); all "breakable" nonring single bonds are broken to generate substituent
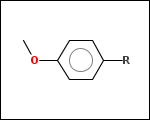
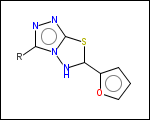
-r2 - spacers (groups with 2 attachment points)
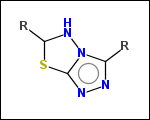
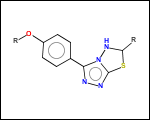
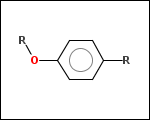
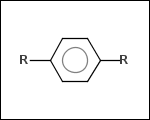
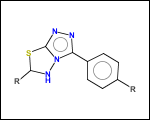
-ringSystems - ring systems is a collection of fused or spiro rings
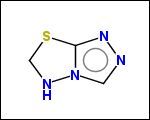
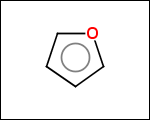
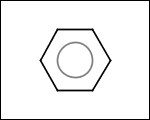
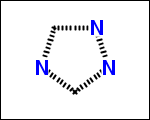
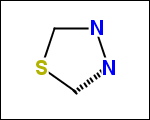
-simpleRings - simple rings which this molecule contains. Simple ring does not need to be a valid molecule (in example below, in the sulfur ring only 2 atoms are aromatic) therefore results are provided as fragment SMILES (note that aromatic bonds in fragments are displayed as dashed lines on images below)
C1Sc:nN1 c:1:n:n:c:n:1 c:1:c:c:c:c:c:1 o:1:c:c:c:c:1
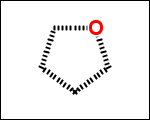

-scaffold - is ring part of a molecule (rings systems and their connections) without aliphatic substituents
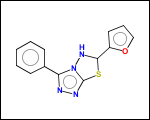
-hose - generates so called HOSE fragments (atoms with environment). A HOSE fragment consists of a central atom (first atom in HOSE SMILES) and several levels of surrounding atoms.
HOSE fragments may be used as structural descriptors by QSAR studies or fragment-based property prediction applications.
Fragmentation parameters
By default, the size of the r1 and r2 fragments is limited to 15 atoms. This may be changed by a parameter -maxsize n (this parameter does not affect other types of fragments).
Generated fragments are written in the output line after input SMILES (in a canonized form) and any other parameters from the input. All items are tab separated.
Parameter -list allows to perform fragmentation statistics for large collections of molecules. On the output a list of fragments is provided, together with the number of molecules containing these fragments.
The following command provides list of substituents up to 8 atoms, which are most common in GPCR ligands.
java -jar mib.jar -f gpcr.smi -standardize -r1 -max 8 -list > gpcr.r1
The first lines of the output file are
[R]C 539 [R]c1ccccc1 213 [R]O 204 [R]OC 193 [R]N 191 [R]Cl 172 [R]F 133 [R]CC 83 [R]CCC 71 [R]C(O)=O 67 . . .
When using parameter -count also the number of fragments of particular type will be provided, in the form fragment1 count1 fragment2 count2 (tab separated)
For example command
java -jar mib.jar -smi 'c1ncccc1' -hose -maxSize 1
provides output
c1ccncc1 [n] [cH]
while when using also the parameters -count the output includes also the number of respective fragments in the molecule
c1ccncc1 [n] 1 [cH] 5
When using the parameter -count together with the parameter -list, the number on output provides the total number of fragments of this type in the molecule set (and not just the number of molecules with this fragments as by -list alone).
-maxsize sets the maximum size of generated fragments. This is ignored when generating rings and scaffolds, and makes sense only by -r1 and -r2 fragments and HOSE fragments (in this case -maxsize is the number of surrounding levels, 1 - just central atom, 2 - single level of neighbors, 3 - 2 levels of neighbors, etc)
Fragmentation is applied only to the main part of multipart molecules (as the keyword -singlepart would be used). [This may be modified in later release of the toolkit].
-tautomer.
In the output line canonized SMILES of the original molecule, followed by eventual data contained in the input line are provided, followed by a number giving the number of generated tautomers and SMILES codes of these tautomers. All data in line are tab separated.
The command
java mib -smi 'n1c(O)cccc1' -tautomers
provides the following output
Oc1ccccn1 2 O=c1cccc[nH]1 Oc1ccccn1
To get tautomers of a single molecule one tautomers SMILES per line (this output may be displayed for example by the Molinspiration molecule viewer) use the -list option.
The command
java -jar mib.jar -smi 'Nc2nc1nc[nH]c1c(=S)[nH]2' -tautomers -list > tautomers.smi
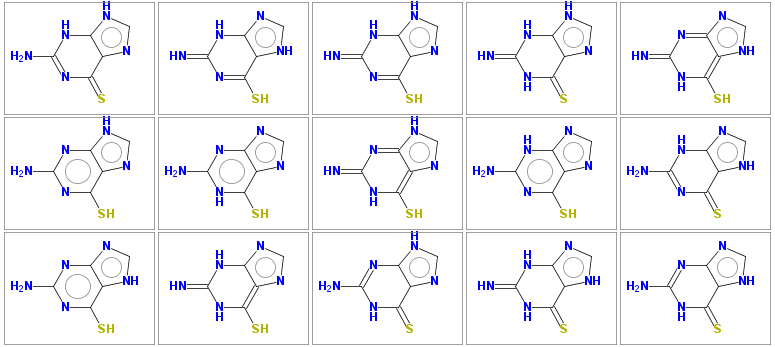
saves the following output to the tautomers.smi file (the source SMILES in this example is thioguanine)
Nc2nc(=S)c1nc[nH]c1[nH]2 1 Sc1nc(=N)[nH]c2nc[nH]c12 1 Sc1nc(=N)[nH]c2[nH]cnc12 1 N=c2[nH]c(=S)c1nc[nH]c1[nH]2 1 Sc1[nH]c(=N)nc2nc[nH]c12 1 Nc2nc(S)c1nc[nH]c1n2 1 Nc2nc1ncnc1c(S)[nH]2 1 Sc1[nH]c(=N)nc2[nH]cnc12 1 Nc2nc(S)c1ncnc1[nH]2 1 Nc2nc(=S)c1[nH]cnc1[nH]2 1 Nc2nc(S)c1[nH]cnc1n2 1 Sc1[nH]c(=N)[nH]c2ncnc12 1 Nc2nc1[nH]cnc1c(=S)[nH]2 1 N=c2[nH]c(=S)c1[nH]cnc1[nH]2 1 Nc2nc1nc[nH]c1c(=S)[nH]2 1
The 15 tautomers generated are shown below
Some molecules may have very large number of tautomers (several hundreds), to keep computational time reasonable, the default number of generated tautomers is limited to 50. This limit may be increased by parameter -maxtautomers n .
Tautomers are listed on the output in alphabetic order.
EZ stereochemistry on tautomeric bonds is not preserved during tautomer enumeration.
Contact us please at info[at]molinspiration.com to arrange an evaluation license of mib.