
|
The miscreen engine first analyzes a training set of active structures (in extreme cases, even a single active molecule may be sufficient to build a usable model) and compares it with inactive molecules using advanced Bayesian statistical methods. Only SMILES representations of active and inactive molecules are required for training; no information about the active site or binding mode is necessary. This is particularly useful in projects where structure-based approaches cannot be applied because information about the receptor's 3D structure is unavailable. Based on this analysis, a fragment-based model is generated in which a bioactivity contribution is assigned to each substructure fragment. Once the model is built, the bioactivity of screened molecules can be calculated as the sum of activity contributions of fragments present in those molecules. This results in a molecular activity score (typically ranging from -3 to 3). Molecules with higher activity scores have a greater probability of exhibiting biological activity. Such in silico screening is highly efficient, allowing screening of molecular collections containing several million compounds within approximately one hour.
The schematic workflow of the Molinspiration virtual screening protocol is shown below.
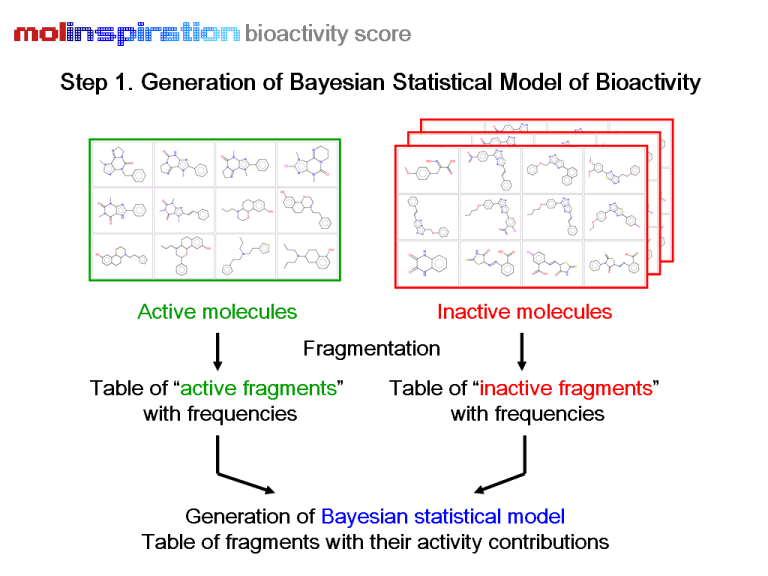
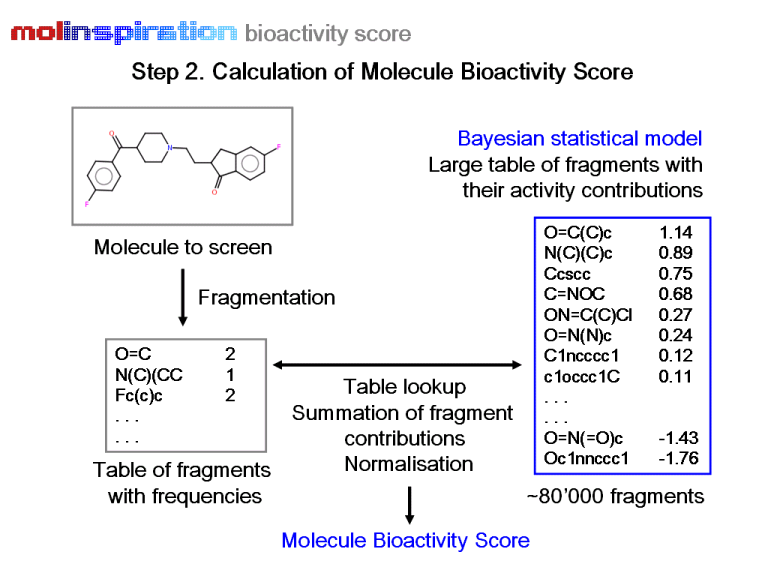
Based on the protocol described above, we have developed ready-to-use screening models for four important drug classes: GPCR ligands, ion channel blockers, kinase inhibitors, and nuclear receptor ligands. A virtual screening model for virtually any target can also be developed easily using the built-in functionality of miscreen. Several of our customers have successfully applied Molinspiration technology in virtual screening and compound purchasing campaigns, design of targeted combinatorial libraries, and have developed and applied their own models for identification of ligands for various GPCR receptors, including chemokine, melanocortin, glutamate, histamine, and serotonin receptors, as well as models for virtual screening of novel pesticides.
Another advantage of our Bayesian statistics-based virtual screening protocol is its ability to generalize by learning broader structural requirements associated with biological activity. Therefore, the identified bioactive molecules are not restricted to structures similar to the training set. The protocol is also capable of identifying entirely new classes of active structures (scaffold hopping).
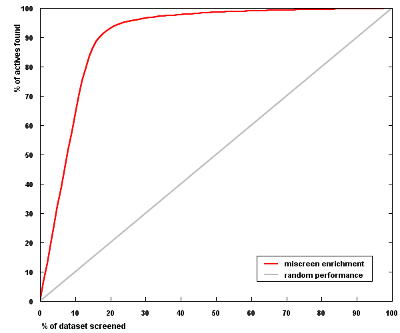
In the validation study below, 21,000 diverse protease inhibitors collected from medicinal chemistry literature were used as active structures, together with approximately 120,000 drug-like organic molecules from the ZINC database as inactive compounds. A cross-validation setup (80% of data used for training and 20% for testing) was employed. The enrichment obtained for the test set is shown in the graph (average of 10 cross-validation runs). Similar results have also been obtained for other activity classes, including GPCR ligands, ion channel modulators, kinase inhibitors, insecticides, fungicides, and herbicides.
If you would like more information about the Molinspiration virtual screening package miscreen, or would like to evaluate the engine at your site, please contact us at info[at]molinspiration.com.
Back to Molinspiration Home